Arvelige arytmisyndromer kalles også primær elektrisk sykdom. Disse er karakterisert av et strukturelt normalt hjerte, og underliggende etiologi er ofte ionekanalsykdommer. Ionekanalene regulerer hjertets depolarisering og repolarisering, og de viktigste sykdommene er Lang QT-tid syndrom (LQTS), Brugada syndrom (BrS) og katekolaminerg polymorf ventrikkeltakykardi (CPVT). De er alle autosomalt dominant arvelige. Dette betyr at gutter og jenter har lik risiko (50 % risiko) for å arve en forelders variant.
Arvelige arytmisyndromer
- Revidert:
- 05.12.2024
- Sist endret:
- 24.06.2026
Forfatter:
Kristina Haugaa
Lang QT-tid syndrom
- Revidert:
- 05.12.2024
- Sist endret:
- 24.06.2026
Forfatter:
Kristina Haugaa
Lang QT-tid syndrom (LQTS) er en genetisk hjertesykdom som karakteriseres av forlenget repolariseringstid, som kan sees som forlenget QT-tid og som disponerer for ventrikulære arytmier som kan gi synkoper eller plutselig hjertedød. Den mest klassiske ventrikulære arytmien er Torsades de Pointes (TdP), en akse-alternerende polymorf ventrikkeltakykardi som opptrer i forbindelse med forlenget QT-tid. Forlenget QT-intervall kan også ses ved en rekke ervervede tilstander, ofte betinget i medisinbruk eller elektrolyttforstyrrelser.
Ventrikulære arytmier preger symptombildet, og pasientene presenterer seg ofte med palpitasjoner, svimmelhet, synkope/nærsynkope eller hjertestans.
LQTS diagnostiseres ved QTc>480 ms, eller ved QTc>460 ms og ventrikulær arytmi eller overlevd hjertestans, eller ved påvist sikkert sykdomsfremkallende genetisk variant (uavhengig av QT-tid). En forlenget QT-tid ved et enkelt EKG kan gi mistanke om LQTS, mens forlenget QT ved gjentatte målinger sammen med symptomer (synkoper, arytmier) taler for LQTS, selv uten sikker sykdomsgivende variant. Genetisk testing bør gjøres ved klar klinisk mistanke om LQTS. Dersom man påviser en variant, åpner dette for muligheten for genetisk kaskadescreening.
LQTS er en ionekanalsykdom der defekte kalium eller natrium-kanaler kan være årsaken til sykdommen. De tre vanligste typene er LQTS type 1 (genvariant i KCNQ1, IKs, langsom K-kanal), LQTS type 2 (genvariant i KCNH2, IKr, rask kaliumkanal) og LQTS type 3 (genvariant i SCN5A, Na-kanal). De ulike typene har litt ulik fenotype, LQT1 får typisk arytmianfall ved fysisk aktivitet og adrenerg stimulus, LQT2 ved plutselige lyder, psykisk stress og i postpartumperioden. LQT3 pasienter får arytmianfall i hvile og søvn.
Medikamentell behandling
Betareseptorantagonister (Adrenerge betareseptorantagonister) anbefales til alle pasienter med en klinisk LQTS-diagnose, og skal også vurderes hos genvariantpositive familiemedlemmer uten symptomer. Betareseptorantagonister bør opptitreres til maksimalt tolererbar dose basert på bivirkninger, ikke pulsmål. Vanlig startdose er 50 mg metoprolol depot som trappes opp. Det er stor variasjon i doserespons og i tolerabilitet. Studier har vist at uselektive betareseptorantagonister som propranolol og nadolol trolig har bedre antiarytmisk effekt og bør være det foretrukne medikamentet. Nadolol er en ikke-selektiv betablokker med lang halveringstid. Nadolol søkes på registreringsfritak og fås på blå resept etter individuell søknad. Ekvipotente doser er 40 mg nadolol-50 mg metoprolol depot.
Dersom pasientene har tilleggslidelser, og det er behov for andre medikamenter som kan gi økt QT-tid (antidepressiva, organtransplantasjon etc.), og det er vanskelig å finne et ikke-QT-forlengende alternativ, kan det være indikasjon for øking eller oppstart av betareseptorantagonist hos dem som ikke er behandlet fra før. Dette må vurderes individuelt. Seksjon for kardiogenetikk, Kardiologisk avdeling OUS, Rikshospitalet har spesialkompetanse på området.
Annen behandling
Livsstilsintervensjoner er sentrale i behandlingen av pasienter med LQTS. Det viktigste er å unngå QT-forlengende medikamenter som er listet på internettdatabasen www.crediblemeds.org. Per i dag er det over 300 medikamenter på denne listen, og denne må konsulteres ved all medikamentell behandling av LQTS-pasienter. Noen hovedgrupper av medikamenter som er mye brukt i vanlig klinisk praksis og som er QT-tid-forlengende er antibiotika (spesielt erytromycin), antidepressiva (bl.a.SSRI som escitalopram, mirtazapin), antipsykotika (bl.a. olanzapin, kvetiapin), alle kvalmestillende (bl.a. ondansetron), og antiarytmika (bl.a. flekainid).
Pasienter med LQTS må være nøye med hydrering og salt/kalium-tilskudd ved mye svetting og gastroenteritt, aldri bade alene, og unngå å drive med konkurranseidrett.
Annen behandling inkluderer ICD-implantasjon (Implanterbar defibrillator (ICD)) og sympatektomi.
Akuttbehandling
- Behandling av symptomatisk TdP og arytmistorm i tillegg til betareseptorantagonistbehandling (Adrenerge betareseptorantagonister)
- Defibrillering ved bevissthetstap
- Intravenøs magnesium
- Tiltak for å opprettholde en relativt rask hjerterytme (100-110 slag/minutt): Isoprenalin-infusjon eller temporær pacing.
- Korreksjon av elektrolyttforstyrrelser. Tilstrebe høy-normal kalium
- Seponere eventuelt QT-forlengende medikasjon
Brugada syndrom
- Revidert:
- 05.12.2024
- Sist endret:
- 24.06.2026
Forfatter:
Kristina Haugaa
Brugada syndrom (BrS) kjennetegnes ved et karakteristisk EKG-mønster med ST-elevasjon i minst én av de høyresidige prekordialledningene (V1-V3) i et strukturelt tilnærmet normalt hjerte. Pasientene har høy risiko for å utvikle potensielt livstruende ventrikulære arytmier. Man får en sikker BrS-diagnose når type-I-EKG-forandringer (Figur 1) observeres enten spontant eller etter intravenøs provokasjon med en natriumkanalblokker i minst én høy prekordial EKG-avledning (V1 og/eller V2) som er plassert i standard posisjon eller i høy posisjon (2. eller 3. interkostalrom).
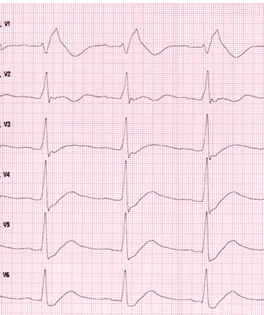
Figur 1: EKG med normofrekvent sinusrytme, tydelige Type 1 Brugada forandringer i V1.
Prevalensen av BrS er usikker og angitt til cirka 1:5 000 til 1:10 000 i Skandinavia, men tilstanden er hyppigere iblant annet Asia. Økt oppmerksomhet rundt sykdommen gjør at den er stadig hyppigere diagnostisert også i Norge.
Genotype positiv BrS skyldes varianter i proteiner som koder for kardiale natriumkanaler (SCN5A), og arvegangen er autosomal dominant. Imidlertid påvises sikker Brugada-relatert mutasjon kun hos rundt 30 % av pasientene.
Foruten de klassiske EKG-forandringene, er det arytmier som dominerer symptombildet. Mest fryktet er polymorf ventrikkeltakykardi og ventrikkelflimmer. Således er synkope/nærsynkope og hjertestans de vanligste symptomene, og ses hos BrS-pasienter oftest i forbindelse med søvn og hvile (lav sympatikotonus). Overoppheting (feber), store matinntak og bruk av visse medisiner kan også utløse arytmier. Tilstanden debuterer typisk hos unge og middelaldrende voksne, og variantpositive menn har 7–8 ganger høyere sykdomspenetrans enn kvinner. Kramper og nattlig agonal respirasjon er også symptomer som kan mistolkes og føre til oversett diagnose.
BrS EKG-mønsteret og arytmirisiko kan bli provosert av mange forskjellige typer medisiner, inkludert antiarytmika, antipsykotika og alkohol. Se medikamentlisten som ligger på nettsiden www.brugadadrugs.org. Pasienter må opplyses om å ha et aktivt forhold til denne nettsiden og informere behandlende leger om dette.
Det finnes ingen profylaktisk medikamentell behandling mot BrS, så ICD (Implanterbar defibrillator (ICD)) er den eneste sikre antiarytmiske behandlingen ved BrS. Det er vanskelig å identifisere de pasientene som trenger ICD (se nedenfor). Noen studier har vist at kinidin kan brukes ved gjentatte ICD-støt, eller hos høyrisikopasienter som nekter ICD eller har andre kontraindikasjoner.
Underkapitler
Arytmistorm ved Brugada syndrom
- Revidert:
- 05.12.2024
- Sist endret:
- 24.06.2026
Forfatter:
Kristina Haugaa
Livstruende arytmier behandles etter vanlige retningslinjer med noen særpreg (se også Hjerterytmeforstyrrelser). Det er flere rapporter om at lavdosert isoprenalin-infusjon har reversert EKG-forandringer og stoppet residiverende arytmier.
- Defibriller om nødvendig, og overfør pasienten til intensivenhet
- Korriger utløsende faktorer:
- Stopp medikamenter som fremmer arytmi
- Behandle feber, eventuelt kjøle pasienten
- Oppretthold normal elektrolyttbalanse
- Antiarytmisk behandling:
- Isoprenalin (1–2 µg bolus i.v. fulgt av infusjon med vektjustert 0.002-0.03 µg/kg/min)
- Kinidin (300–1500 mg/dag). Mål for kinidin plasma-konsentrasjon: 1-3 µg/mL eller 3,5-11 µmol/L. Noen pasienter trenger kun lave doser (f.eks. ≤ 600 mg daglig). Barn doseres etter kroppsvekt, og 30-60 mg/kg/dag fordelt på 4 doser har vært anbefalt.
Legemidler
Katekolaminerg polymorf ventrikkeltakykardi (CPVT)
- Revidert:
- 11.03.2025
- Sist endret:
- 24.06.2026
Forfatter:
Kristina Haugaa
Katekolaminerg polymorf ventrikkeltakykardi (CPVT) er en arvelig hjertesykdom preget av besvimelser og risiko for plutselig død ved fysisk aktivitet eller stress. Diagnosen baseres på stressindusert ventrikkeltakykardi i et normalt strukturelt hjerte eller ved genetisk testing. Prevalensen anslås til omtrent 1:10 000 og er likt fordelt mellom kvinner og menn. CPVT skyldes genvarianter som påvirker kalsiumreguleringen i hjertet, hovedsakelig i genet for ryanodinreseptor 2 (RyR2) som arves autosomalt dominant.
Symptomer inkluderer besvimelser under fysisk aktivitet eller stress, og plutselig hjertedød kan være første presentasjon av sykdommen. Som hos LQTS-pasienter er svømming, særlig i kaldt vann, en klassisk utløsende faktor for arytmi, og man bør tenke på ionekanalsykdommer ved drukningsulykker hos svømmedyktige barn og unge.
Diagnosen CPVT kan være vanskelig å stille, fordi hvile-EKG, senpotensial-EKG, elektrofysiologiske undersøkelser, ekkokardiografi og klinisk undersøkelse ofte er upåfallende. Diagnosen stilles oftest ved arbeids-EKG, der man påviser ventrikulære arytmier av økende alvorlighetsgrad med økende hjertefrekvens og arbeidsintensitet.
Genetisk testing bør utføres ved sterk klinisk mistanke om sykdommen. Dersom man påviser en sykdomsgivende variant, bør førestegradsslektninger testes for den samme varianten (genetisk kaskadescreening), og variantpositive familiemedlemmer må tilbys forebyggende behandling og følges opp av kardiolog med kompetanse på området.
Behandling innebærer livsstilsendringer for å unngå triggere, som å unngå konkurranseidrett og å bade alene. I tillegg bør man forsøke å unngå situasjoner med mye psykisk stress.
Betablokkere er standard medikamentell behandling hos CPVT-pasienter, og man anbefaler også betablokkere hos asymptomatiske mutasjonspositive familiemedlemmer fordi det er høy penetrans av sykdommen og plutselig hjertedød kan vært første presentasjon. Det er helt essensielt at pasienten husker å ta tablettene hver dag; ofte ser man at CPVT-dødsfall kan kobles opp til glemte medikamentdoser. Norske studier har vist bedre arytmikontroll ved arbeids-EKG under behandling med nadolol sammenliknet med selektive betablokkere og i dag er de europeiske anbefalinger at CPVT-pasientene skal behandles med nadolol som førstehåndspreparat. Nadolol søkes på registreringsfritak og med individuell søknad for legemidler som ikke står på refusjonslisten.
Hvis betablokker ikke gir tilstrekkelig arytmikontroll, er det aktuelt med tilleggsbehandling med flekainid. Flekainid gis da i kombinasjon med betablokker i uendret dose.
ICD anbefales for dem med alvorlige arytmier til tross for medikamentell behandling.
Prognosen varierer, men mange opplever lite arytmier når de er under behandling. Pasienter som er diagnostisert i voksen alder, som ikke har arytmier ved arbeids-EKG under betablokkerbehandling, og som husker å ta medisinene sine, har trolig god prognose.
Kilder (Arvelige arytmisyndromer)
- Sist endret:
- 24.06.2026
Forfatter:
Kristina Haugaa
Haugaa KH, Leren IS, Berge KE, Bathen J, Loennechen JP, Anfinsen OG, Fruh A, Edvardsen T, Kongsgard E, Leren TP, et al. High prevalence of exercise-induced arrhythmias in catecholaminergic polymorphic ventricular tachycardia mutation-positive family members diagnosed by cascade genetic screening. Europace : European pacing, arrhythmias, and cardiac electrophysiology : journal of the working groups on cardiac pacing, arrhythmias, and cardiac cellular electrophysiology of the European Society of Cardiology. 2010;12:417-423. doi: 10.1093/europace/eup448
Leren IS, Haugaa KH, Edvardsen T, Anfinsen OG, Kongsgard E, Berge KE, Leren TP, Amlie JP. Catecholaminergic polymorphic ventricular tachycardia. Tidsskrift for den Norske laegeforening : tidsskrift for praktisk medicin, ny raekke. 2010;130:139-142. doi: 10.4045/tidsskr.09.0529
Leren IS, Saberniak J, Majid E, Haland TF, Edvardsen T, Haugaa KH. Nadolol decreases the incidence and severity of ventricular arrhythmias during exercise stress testing compared with beta1-selective beta-blockers in patients with catecholaminergic polymorphic ventricular tachycardia. Heart rhythm. 2016;13:433-440. doi: 10.1016/j.hrthm.2015.09.029
Pflaumer A, Davis AM. Guidelines for the diagnosis and management of Catecholaminergic Polymorphic Ventricular Tachycardia. Heart Lung Circ. 2012;21:96-100. doi: 10.1016/j.hlc.2011.10.008
Watanabe H, Minamino T. Genetics of Brugada syndrome. J Hum Genet, 2016. 61(1): 57-60. 3.
Wilde AA, Antzelevitch C, Borggrefe M, Brugada J, Brugada R, Brugada P, Corrado D, Hauer RN, Kass RS, Nademanee K, Priori SG, Towbin JA. Proposed diagnostic criteria for the Brugada syndrome: consensus report. Circulation, 2002. 106(19): 2514-2519.
2.
Zeppenfeld K, Tfelt-Hansen J, de Riva M, Winkel BG, Behr ER, Blom NA, Charron P, Corrado D, Dagres N, de Chillou C, Eckardt L, Friede T, Haugaa KH, Hocini M, Lambiase PD, Marijon E, Merino JL, Peichl P, Priori SG, Reichlin T, Schulz-Menger J, Sticherling C, Tzeis S, Verstrael A, Volterrani M. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J, 2022. 43(40): 3997-4126.